[ad_1]
The AlphaFold method
Many novel machine learning innovations contribute to AlphaFold’s current level of accuracy. We give a high-level overview of the system below; for a technical description of the network architecture see our AlphaFold methods paper and especially its extensive Supplementary Information.
The AlphaFold network consists of two main stages. Stage 1 takes as input the amino acid sequence and a multiple sequence alignment (MSA). Its goal is to learn a rich “pairwise representation” that is informative about which residue pairs are close in 3D space.
Stage 2 uses this representation to directly produce atomic coordinates by treating each residue as a separate object, predicting the rotation and translation necessary to place each residue, and ultimately assembling a structured chain. The design of the network draws on our intuitions about protein physics and geometry, for example, in the form of the updates applied and in the choice of loss.
Interestingly, we can produce a 3D structure based on the representation at intermediate layers of the network. The resulting “trajectory” videos show how AlphaFold’s belief about the correct structure develops during inference, layer by layer. Typically a hypothesis emerges after the first few layers followed by a lengthy process of refinement, although some targets require the full depth of the network to arrive at a good prediction.
Accuracy and confidence
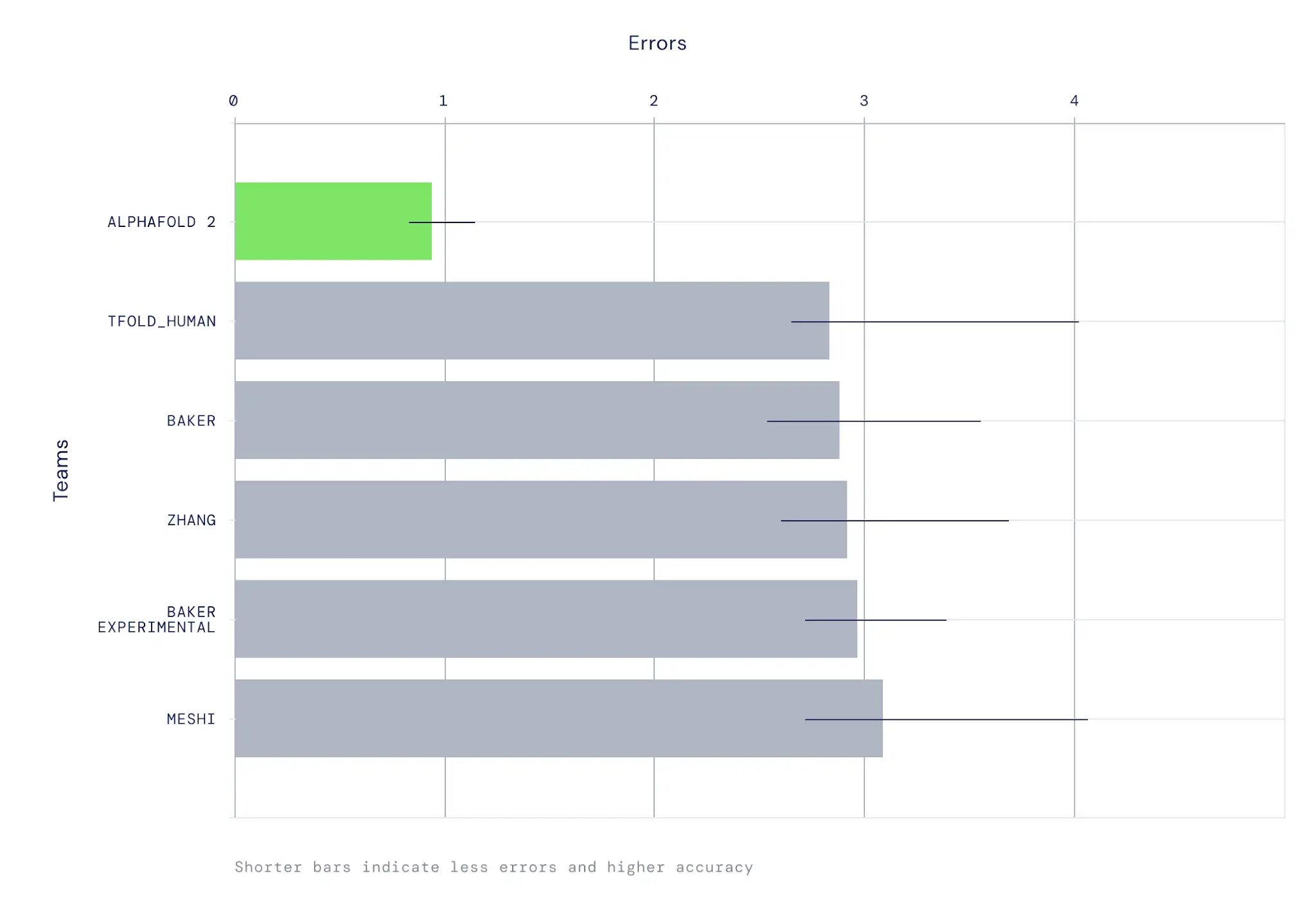
AlphaFold was stringently assessed in the CASP14 experiment, in which participants blindly predict protein structures that have been solved but not yet made public. The method achieved high accuracy in a majority of cases, with an average 95% RMSD-Cα to the experimental structure of less than 1Å. In our papers, we further evaluate the model on a much larger set of recent PDB entries. Among the findings are strong performance on large proteins and good side chain accuracy where the backbone is well-predicted.

An important factor in the utility of structure predictions is the quality of the associated confidence measures. Can the model identify the parts of its prediction likely to be reliable? We have developed two confidence measures on top of the AlphaFold network to address this question.
The first is pLDDT (predicted lDDT-Cα), a per-residue measure of local confidence on a scale from 0 – 100. pLDDT can vary dramatically along a chain, enabling the model to express high confidence on structured domains but low confidence on the linkers between them, for example. In our paper, we present evidence that some regions with low pLDDT may be unstructured in isolation; either intrinsically disordered or structured only in the context of a larger complex. Regions with pLDDT < 50 should not be interpreted except as a possible disorder prediction.
The second metric is PAE (Predicted Aligned Error), which reports AlphaFold’s expected position error at residue x, when the predicted and true structures are aligned on residue y. This is useful for assessing confidence in global features, especially domain packing. For residues x and y drawn from two different domains, a consistently low PAE at (x, y) suggests AlphaFold is confident about the relative domain positions. Consistently high PAE at (x, y) suggests the relative positions of the domains should not be interpreted. The general approach used to produce PAE can be adapted to predict a variety of superposition-based metrics, including TM-score and GDT.
.jpg)
To emphasise, AlphaFold models are ultimately predictions: while often highly accurate they will sometimes be in error. Predicted atomic coordinates should be interpreted carefully, and in the context of these confidence measures.
Open sourcing
Alongside our method paper, we have made the AlphaFold source code available on GitHub. This includes access to a trained model and a script for making predictions on novel input sequences. We believe this is an important step that will enable the community to use and build on our work. The easiest way to fold a single new protein with AlphaFold is to use our Colab notebook.
The open source code is an updated version of our CASP14 system based on the JAX framework, and it achieves equally high accuracy. It also incorporates some recent performance improvements. AlphaFold’s speed has always depended heavily on the input sequence length, with short proteins taking minutes to process and only very long proteins running into hours. Once the MSA has been assembled, the open source version can now predict the structure of a 400 residue protein in just over a minute of GPU time on a V100.
Proteome scale and AlphaFold DB
AlphaFold’s fast inference times allow the method to be applied at whole-proteome scale. In our paper, we discuss AlphaFold’s predictions for the human proteome. However, we have since generated predictions for the reference proteomes of a number of model organisms, pathogens and economically significant species, and large scale prediction is now routine. Interestingly, we observe a difference in the pLDDT distribution between species, with generally higher confidence on bacteria and archaea and lower confidence on eukaryotes, which we hypothesize may be related to the prevalence of disorder in these proteomes.
No single research group can fully explore such a large dataset, and so we partnered with EMBL-EBI to make the predictions freely available via the AlphaFold DB. Each prediction can be viewed alongside the confidence metrics described above. A bulk download is also provided for each species, and all data is covered by a CC-BY-4.0 license (making it freely available for both academic and commercial use). We are extremely grateful to EMBL-EBI for their work with us to develop this new resource. Over the course of the coming months we plan to expand the dataset to cover the over 100 million proteins in UniRef90.
.jpg)
.jpg)
In AlphaFold DB, we have chosen to share predictions of full protein chains up to 2700 amino acids in length, rather than cropping to individual domains. The rationale is that this avoids missing structured regions that have yet to be annotated. It also provides context from the full amino acid sequence, and allows the model to attempt a domain packing prediction. AlphaFold’s intra-domain accuracy was more extensively evaluated in CASP14 and is expected to be higher than its inter-domain accuracy. However, AlphaFold was the top ranked method in the inter-domain assessment, and we expect it to produce an informative prediction in some cases. We encourage users to view the PAE plot to determine whether domain placement is likely to be meaningful.
Future work
We are excited about the future for computational structural biology. There remain many important topics to address: predicting the structure of complexes, incorporating non-protein components, and capturing dynamics and the response to point mutations. The development of network architectures like AlphaFold that excel at the task of understanding protein structure is a cause for optimism that we can make progress on related problems.
We see AlphaFold as a complementary technology to experimental structural biology. This is perhaps best illustrated by its role in helping to solve experimental structures, through molecular replacement and docking into cryo-EM volumes. Both applications can accelerate existing research, saving months of effort. From a bioinformatics perspective, AlphaFold’s speed enables the generation of predicted structures on a massive scale. This has the potential to unlock new avenues of research, by supporting structural investigations of the contents of large sequence databases.
Ultimately, we hope AlphaFold will prove a useful tool for illuminating protein space, and we look forward to seeing how it is applied in the coming months and years.
We would love to hear your feedback and understand how AlphaFold and the AlphaFold DB have been useful in your research. Share your stories at alphafold@deepmind.com.
[ad_2]
Source link